Category
page 1Computational chemistry

computational chemistry
branch of chemistry

energy level
different states of quantum systems
Hamiltonian operator
quantum operator for the energy
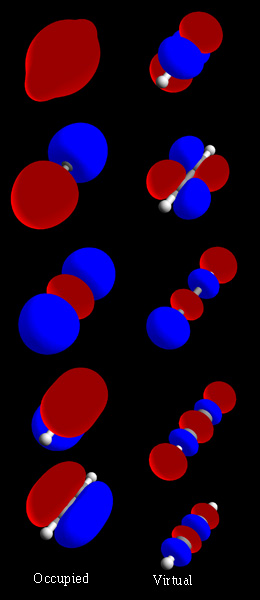
molecular orbital
wave-like behavior of an electron in a molecule

Folding@home
Folding@home (FAH or F@h) is a distributed computing project aimed to help scientists develop new therapeutics for a variety of diseases by the means of simulating protein dynamics. This includes the process of protein folding and the movements of proteins, and is reliant on simulations run on volunteers' personal computers. Folding@home is currently based at the University of Pennsylvania and led by Greg Bowman, a former student of Vijay Pande.
molecular dynamics simulation
method of computer simulation of molecular interaction
cheminformatics
Cheminformatics (also known as chemoinformatics) refers to the use of physical chemistry theory with computer and information science techniques—so called "in silico" techniques—in application to a range of descriptive and prescriptive problems in the field of chemistry, including in its applications to biology and related molecular fields. Such in silico techniques are used, for example, by pharmaceutical companies and in academic settings to aid and inform the process of drug discovery, for instance in the design of well-defined combinatorial libraries of synthetic compounds, or to assist in
zanamivir
Zanamivir, sold under the brand name Relenza among others, is an anti-viral medication used to treat and prevent influenza caused by influenza A and influenza B viruses. It is a neuraminidase inhibitor and was developed by the Australian biotech firm Biota Holdings. It was licensed to Glaxo Wellcome in 1990 and approved in the US in 1999, only for use as a treatment for influenza. In 2006, it was approved for prevention of influenza A and B. Zanamivir is the first neuraminidase inhibitor commercially developed. It was developed by GlaxoSmithKline.
perturbation theory
mathematical methods used to find an approximate solution to a problem which cannot be solved exactly
Lennard-Jones potential
mathematical model that approximates the interaction between a pair of neutral atoms or molecules
chemometrics
Chemometrics is the science of extracting information from chemical systems by data-driven means. Chemometrics is inherently interdisciplinary, using methods frequently employed in core data-analytic disciplines such as multivariate statistics, applied mathematics, and computer science, in order to address problems in chemistry, biochemistry, medicine, biology and chemical engineering. In this way, it mirrors other interdisciplinary fields, such as psychometrics and econometrics.
molecular modelling
discovering chemical properties by physical simulations
quantitative structure-activity relationship
quantitative prediction of the biological, ecotoxicological or pharmaceutical activity of a molecule
Hartree–Fock method
method of approximation for the determination of the wave function and the energy of a quantum many-body system in a stationary state
molecular docking
attempt to predict the structure of the intermolecular complex formed between two or more molecules
molecular mechanics
use of classical mechanics to model molecular systems
reaction coordinate
abstract coordinate depicting chemical reaction progress
Slater determinant
expression that describes the wave function of a multi-fermionic system
Variational method
Method for the determination of the approximate ground state of a quantum mechanical system.

chemogenomics
thumb|300px|Chemogenomics Stäubli|Staubli robot retrieves assay plates from incubators
chemical database
database specifically designed to store chemical information

Critical Assessment of protein Structure Prediction
thumb|right|250px|A target structure (ribbons) and 354 template-based predictions superimposed (gray Calpha backbones); from CASP8
Critical Assessment of Structure Prediction (CASP), sometimes called Critical Assessment of Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and sof
Ab initio quantum chemistry methods
category of computational quantum chemistry technique
Basis set
chemical term

Koopmans' theorem
theorem in quantum mechanics that in closed-shell Hartree–Fock theory, the first ionization energy of a molecular system equals the negative of the orbital energy of the highest occupied molecular orbital
Q1069211
markup language and file format
semiclassical physics
physical model treating some aspects in terms of quantum mechanics and others by classical physics
partial charge
Phi coefficient
type of coefficient
Fermi resonance
shift of energy in physics
water model
model to simulate effects of water in computational chemistry,
Ewald summation
Computation method named after Paul Peter Ewald

Gaussian orbital
Mathematical function
Møller–Plesset perturbation theory
computational chemistry ab initio method that adds to the Hartree–Fock method electron correlation effects using Rayleigh–Schrödinger perturbation theory
Bette Korber
American computational biologist
Slater-type orbital
function used to describe atomic orbitals in quantum chemistry
Post-Hartree–Fock
In computational chemistry, post–Hartree–Fock (post–HF) methods are the set of methods developed to improve on the Hartree–Fock (HF), or self-consistent field (SCF), method. They add electron correlation which is a more accurate way of including the repulsions between electrons than in the Hartree–Fock method where repulsions are only averaged.
self-avoiding walk
a sequence of moves on a lattice that does not visit the same point more than once
isodesmic reaction

Molecule Editor
Software used to edit molecular structures computationally.
Journal of Chemical Information and Modeling
peer-reviewed scientific journal
Journal of Chemical Theory and Computation
journal
Buckingham potential
interatomic potential energy model
chemical space
property space of all possible molecules/compounds within given boundary conditions
Z-matrix
molecular modeling tool in chemistry
diffusion Monte Carlo
quantum Monte Carlo method that uses a Green's function to solve the Schrödinger equation
Embedded atom model
Energy profile
representation of a chemical process as a single energetic pathway
Protein Data Bank
textual file format describing the three-dimensional structures of molecules held in the Protein Data Bank
Hartree equation
equation in solid state physics
COSMO solvation model
computational model for solvent effects
Coulomb operator
quantum mechanical operator used in the field of quantum chemistry
QM/MM
The hybrid QM/MM (quantum mechanics/molecular mechanics) approach is a molecular simulation method that combines the strengths of ab initio QM calculations (accuracy) and MM (speed) approaches, thus allowing for the study of chemical processes in solution and in proteins. The QM/MM approach was introduced in the 1976 paper of Warshel and Levitt. They, along with Martin Karplus, won the 2013 Nobel Prize in Chemistry for "the development of multiscale models for complex chemical systems".
time-dependent density functional theory
quantum mechanical theory to investigate the properties and dynamics of many-body systems in the presence of time-dependent potentials